Projects
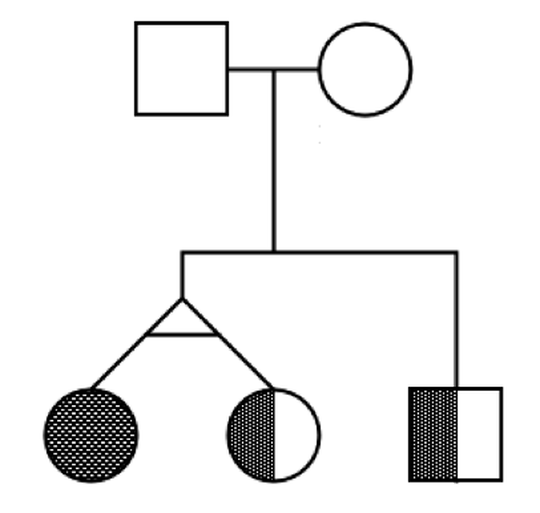
Cholesteatoma - rare variant discovery
Cholesteatoma is a rare disease that results in cyst-like growths in the inner ear. In order to gain further understanding into the aetiology of the disease, nine family’s exomes were sequenced. Here we show some of the analyses run as part of our germline variant discovery pipeline.
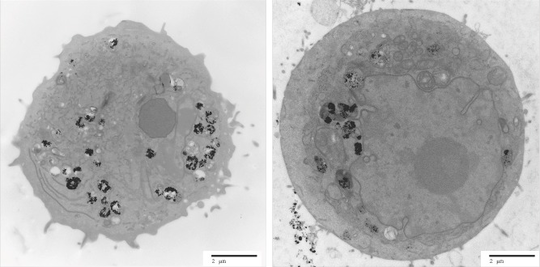
Colorectal cancer tumoroid project
This project utilised DNA-exome sequencing on colorectal tumoroid and normal organoid cultures grown in 3D culture and were analysed for somatic mutations to identify patient specific drug targets. Here we show some of the analyses run as part of our Exome and somatic variant discovery pipeline.